
Cornelia de Lange Syndrome
Overview
Other Names & Coding
Q87.1, Congenital malformation syndromes predominantly associated with short stature
See ICD-10 for Congenital Malformation Syndromes Associated with Short Stature (icd10data.com) for further coding details.
Prevalence
Genetics
Gene mutations causing CdLS affect proteins originally identified to be involved in cohesion of sister chromatids (sister chromatids are chromatids located on the same chromosome). More recently, these proteins have also been shown to regulate gene expression and transcription. CdLS is considered to be one of the emerging class of disorders called "disorders of transcription regulation," and some of the clinical findings overlap with those in CdLS and other disorders with mutations in cohesin. [Olley: 2018] [Parenti: 2016] [Dorsett: 2009] [McNairn: 2008] [Liu: 2009] [Deardorff: 2012] [Deardorff: 2012]
Prognosis
Practice Guidelines
Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, Ishman SL, Kerr LM, Levin AV, Mulder PA, Ramos FJ,
Wierzba J, Ajmone PF, Axtell D, Blagowidow N, Cereda A, Costantino A, Cormier-Daire V, FitzPatrick D, Grados M, Groves L,
Guthrie W, Huisman S, Kaiser FJ, Koekkoek G, Levis M, Mariani M, McCleery JP, Menke LA, Metrena A, O'Connor J, Oliver C, Pie
J, Piening S, Potter CJ, Quaglio AL, Redeker E, Richman D, Rigamonti C, Shi A, Tümer Z, Van Balkom IDC, Hennekam RC.
Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement.
Nat Rev Genet.
2018.
PubMed abstract
Roles of the Medical Home
Children with CdLS and their families can access specialists familiar with the syndrome at various CdLS Foundation events, including the Biennial Conference (CdLS Foundation). The foundation also supports CdLS Multidisciplinary Clinics (CdLS Foundation) in Philadelphia, and a multidisciplinary clinic for adolescents and adults held in Maryland, a clinic in St Louis, MO, and a clinic through the Shriner’s Hospital in Salt Lake City, Utah. Transitioning the child from pediatric care and the school system to the adult world should begin by age 12. Plans for job training and work placement should begin early in high school so they are in place when needed.
Clinical Assessment
Overview
- Growth
- Development
- Speech/language
- Hearing, at diagnosis and every few years
- Vision, at diagnosis and annually
- Gastrointestinal, for malformation risk and gastroesophageal reflux disease (GERD)
- Genetics, particularly if parents are contemplating additional children
- Dental
- Behavior
Pearls & Alerts for Assessment
Gastroesophageal reflux is nearly always presentConsider an evaluation if there are frequent episodes of distress, irritability, and/or arching. Other signs of GERD include vomiting, weight loss or failure to gain expected weight, food refusal, dysphagia (difficulty swallowing), and/or hematemesis (vomiting of blood). It is recommended to screen everyone for this at diagnosis.
Consider obstructionBecause intestinal volvulus is relatively common in children with CdLS, acute abdominal pain, abdominal distention, and/or bilious emesis should prompt immediate evaluation for obstruction. If a child is gastrostomy tube dependent and there are significant feeding difficulties or high-volume gastric fluid output, obstruction should also be considered. Families should be given information about this potential problem.
Sudden changes in behavior or unusual body movements may be a sign of refluxThe appearance or recurrence of severe esophageal reflux or reflux esophagitis may cause changes in behavior such as crying, an increase in self-injurious behavior, refusal to eat, or unusual posturing movements of the head and neck (Sandifer syndrome).
Decreased but predictable growthChildren with CdLS have decreased growth but should follow a
predictable pattern. Plotting height, weight, and head circumference on
CdLS-specific growth charts (see Developmental Skills Chart (CdLS Foundation) ( 98 KB)) may be helpful.
If children stray markedly from their usual pattern or these charts,
consider other problems, such as decreased thyroid or growth hormone
levels.
98 KB)) may be helpful.
If children stray markedly from their usual pattern or these charts,
consider other problems, such as decreased thyroid or growth hormone
levels.
Middle ear effusion and sensorineural hearing loss are both observed. Consider an ENT evaluation if a middle ear effusion is present since this may be causing hearing loss in children who already have delays in speech and language.
Screening
For Complications
- Gene testing at diagnosis to confirm the molecular basis should be considered
- Echocardiogram
- Renal ultrasound
- Upper GI barium study to rule out malrotation and to assess for GERD
- Audiology assessment
- Ophthalmologic examination
Presentations
|
|
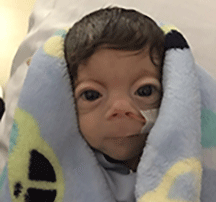


Diagnostic Criteria
- Positive pathogenic mutation on CdLS gene testing, OR
- Nine or more points of which at least 2 are from the cardinal features (facial findings, diaphragmatic hernia, or oligodactyly) and the rest from either cardinal or suggestive features (developmental delay, growth retardation, microcephaly, small hands and/or feet, hirsutism, and brachydactyly 5th finger)
- Synophrys (eyebrows that meet at the midline) and/or thick eyebrows
- Long eyelashes
- Short nose with concave nasal ridge (area and upturned nasal tip)
- Long, prominent, protruding or smooth philtrum (area between upper lip and nose)
- Broad or depressed nasal bridge
- Small chin, may appear more square in adolescence or adulthood
- Thin upper lip, or down-turned corners of mouth
- High or cleft palate
- Widely spaced or absent teeth
- Growth: 2 or more of the following:
- Weight below 5th percentile for age
- Height/length below 5th percentile for age
- Head circumference below 5th percentile for age
- Development: 1 or more of the following:
- Developmental delay or intellectual disability
- Learning disabilities
- Behavior: 2 or more of the following:
- Attention deficit disorder with hyperactivity or constant roaming
- Obsessive-compulsive characteristics
- Anxiety
- Aggression
- Self-injurious behavior
- Extreme shyness or withdrawal
- Autistic features
- Musculoskeletal:
- Limb deficiencies with missing hands or forearms
- Small hands and feet and/or oligodactyly (missing digits)
- 5th finger clinodactyly (curved 5th finger)
- Abnormal palmar crease
- Radial head dislocation/abnormal elbow extension
- Short 1st metacarpal/proximally placed thumb
- Bunion
- Partial 2,3 syndactyly (webbing between 2nd and 3rd toes)
- Scoliosis (curvature of the spine)
- Pectus excavatum (chest or sternum deformity)
- Hip dislocation or dysplasia
- Neurosensory/Skin:
- Ptosis (droopy eyelid)
- Tear duct malformation or blepharitis (inflammation of eyelid)
- Myopia (-6.00 D or more)
- Major eye malformation or peripapillary anomaly
- Deafness or hearing loss
- Seizures
- Cutis marmorata (mottled appearance to skin)
- Hirsutism, generalized (excessive body hair)
- Small nipples and/or umbilicus
- Gastrointestinal malformation/malrotation (approximately 10% of children with CdLS syndrome ) [Deardorff: 2016]
- Diaphragmatic hernia (approximately 1% of children with CdLS syndrome ) [Deardorff: 2016]
- Gastroesophageal reflux disease or GERD (majority of children with CdLS syndrome ) [Deardorff: 2016]
- Cleft palate or submucous cleft palate
- Micropenis
- Hypospadias (abnormally placed opening of urethra on penis)
- Cryptorchidism (undescended testes)
- Renal or urinary tract malformation (approximately 12% of children with CdLS syndrome ) [Deardorff: 2016]
Differential Diagnosis
- Upper limb deformities are present in children with Roberts syndrome.
- Hirsutism and small stature are seen in individuals with Rubinstein-Taybi syndrome.
- Developmental, behavioral, and growth abnormalities are observed in children with fetal alcohol syndrome.
- Developmental delay, behavioral disturbance, and sleep dysfunction are typically present in children with Smith-Magenis syndrome.
- Microcephaly and autism-like features are observed in girls with Rett syndrome.
- Growth restriction is observed in many syndromes.
History & Examination
Current & Past Medical History
Family History
Pregnancy/Perinatal History
Developmental & Educational Progress
98 KB) for expected developmental milestones for children with CdLS.
Social & Family Functioning
Physical Exam
Some of the more common physical exam characteristics are listed below. There are many more that are less common. Children with CdLS may also have various cardiac abnormalities, renal and genital malformations, gastrointestinal malformations, and diaphragmatic hernia.
General
Growth Parameters
Weight, height/length, and head circumference below 5th percentile for
age. See Growth Charts for Boys (CdLS Foundation) ( 874 KB) and Growth Charts for Girls (CdLS Foundation) ( 1.4 MB).
HEENT/Oral
Look for synophrys, long eyelashes, short nose, upturned nasal tip, long and/or smooth philtrum, broad nasal bridge, concave nasal ridge, small chin, thin upper lip, with downturned corners, widely spaced or absent teeth, high or cleft palate, ptosis, myopia, blepharitis. Strabismus, nystagmus, and abnormal gaze behaviors are sometimes seen. Look for middle ear fluid. Sinusitis and nasal polyps may occur. Abnormalities of the inner ear may be observed on CT scan of temporal bone. Look for caries, enamel erosion, and discolored teeth as well as missing teeth, delayed tooth eruption, and delayed loss of primary teeth.
Genitalia
Extremities/Musculoskeletal
Upper limb deformities (significant in approximately 25%), small hands and feet, clinodactyly, abnormal palmar creases, incomplete extension of elbows, and webbing between 2nd and 3rd toes are often present. Assess for scoliosis and dislocated hips. Evaluate for contractures, especially ankles.
Testing
Sensory Testing
Other Testing
- Evaluate or re-evaluate for GERD, when indicated, with EGD or esophageal pH probe. Evaluation information can be found at Gastroesophageal Reflux Disease.
- Consider dual-energy X-ray absorptiometry (DXA) or bone densitometry for osteoporosis in adolescents with CdLS. See Osteoporosis and Pathologic Fractures.
- Consider a modified barium swallow study (MBS) to rule out malrotation, as well as primary or secondary aspiration, in children with poor weight gain, oral aversion, coughing or choking with eating, and/or other feeding difficulties.
- Speech/language assessments periodically
Specialty Collaborations & Other Services
Medical Genetics (see NW providers [1])
Pediatric Gastroenterology (see NW providers [0])
Developmental - Behavioral Pediatrics (see NW providers [1])
Treatment & Management
Pearls & Alerts for Treatment & Management
Provide critical care informationProvide family with information for care in the event of a medical emergency. Include a medical alert card regarding sometimes misdiagnosed problems in children with CdLS. See Critical Care Information (CdLS Foundation) for more information and a printable medical alert card.
Importance of speech/language therapyMost individuals with CdLS, even those that develop the ability to speak in sentences, are delayed in speech-language acquisition; consider early speech/language therapy.
Growth hormoneFamilies may ask about the use of growth hormone for their child's small stature. Growth hormone has not been shown to be effective in CdLS except in children who also have growth hormone deficiency. See Growth Hormone in Children with CdLS (CdLS Foundation).
How should common problems be managed differently in children with Cornelia de Lange Syndrome?
Growth or Weight Gain
874 KB) and Growth Charts for Girls (CdLS Foundation) ( 1.4 MB).
Development (Cognitive, Motor, Language, Social-Emotional)
Common Complaints
Other
Systems
Development (general)
See Augmentative Communication (AAC) and the Developmental Skills Chart (CdLS Foundation) (
98 KB) for
expected age ranges for common developmental milestones. The Portal's module
on Hearing Loss and Deafness provides further management
information.
Specialty Collaborations & Other Services
Developmental - Behavioral Pediatrics (see NW providers [1])
Special Education/Schools (see NW providers [3])
Speech - Language Pathologists (see NW providers [4])
Early Intervention for Children with Disabilities/Delays (see NW providers [3])
Cornelia de Lange Syndrome Clinics (see NW providers [0])
Gastro-Intestinal & Bowel Function
- If GERD is present, medical treatment (H2 blockers, proton pump inhibitors, prokinetics or surgical treatment (Nissen fundoplication and possibly placement of a gastrostomy tube)) is usually necessary. For treatment information, see Gastroesophageal Reflux Disease.
- In addition to short-term pain from reflux esophagitis, long-term esophagitis may lead to a precancerous condition called Barrett's esophagus. Eosinophilic esophagitis may require further evaluation (food allergy testing) and treatment.
- At diagnosis, an upper GI study should be done to rule out malrotation; repair is indicated if present.
- Acute abdominal pain should prompt evaluation for malrotation and volvulus, which may be fatal. Families should have a Medical Alert Card (CdLS Foundation)) to alert clinicians to this possibility.
Specialty Collaborations & Other Services
Pediatric Gastroenterology (see NW providers [0])
Nutrition/Growth/Bone
874 KB)
Growth Charts for Girls (CdLS Foundation) ( 1.4 MB)) to assure growth is appropriate. Nutrition services may be helpful if weight gain is less than expected.Feeding difficulties occur in up to 70% of children with CdLS and may result in not meeting weight gain expectations. Problems may include immature feeding/swallowing patterns, gastroesophageal reflux, or adverse reactions to oral feeding following a period of tube feeding, as well as slow bowel motility. Consultation with a gastroenterologist may be helpful in deciding whether the child may be helped by increasing caloric intake, power packing (see Missing issue with id: 90b0ad6d.xml), or a gastrostomy tube (G-tube) (see Feeding & Nutrition and Feeding Tubes & Gastrostomies in Children). As there are often emotional issues surrounding G-tube insertion, advise families that they can be temporary, can be removed fairly easily (especially percutaneous ones), and that children may usually still be fed orally (unless aspiration is present) even if tube feeding is used.
G-tubes come in several different types, including percutaneous endoscopic gastrostomy tubes (PEGs). PEG insertion usually requires a shorter hospital stay than surgically placed tubes. Parents should understand that G-tubes do not stop gastroesophageal reflux and, in a small percentage of patients, GERD may worsen. G-tubes will not prevent aspiration of secretions, liquids, or solids fed by mouth.
Osteoporosis is also observed frequently. Consider referral to endocrinology for children with fractures and osteoporosis on bone densitometry studies (DXA scan) to consider bisphosphonate treatment. This is particularly important if they are on medications that may increase bone loss, such as seizure medications (e.g., valproic acid), or proton pump inhibitors.
Specialty Collaborations & Other Services
Dieticians and Nutritionists (see NW providers [1])
Pediatric Gastroenterology (see NW providers [0])
Pediatric Endocrinology (see NW providers [1])
Pediatric Orthopedics (see NW providers [4])
Mental Health/Behavior
When dealing with a particular behavior problem, the medical home should first rule out causes of physical discomfort, including gastroesophageal reflux, dental problems, ear and sinus infections, and constipation. Sensory neuropathy may cause painful or tingling sensations, numbness, or insensitivity to pain, and it may present behaviorally if the child has no way to communicate. Discolored hands and feet may be due to Raynaud syndrome, which may cause pain. Specific psychiatric disorders may become evident in adolescence, including depression, obsessive-compulsive disorder, anxiety, and aggression or opposition. (The Portal's module on Depression provides further diagnosis and management information for this condition.)
Once a medical cause for a new behavior is ruled out, obtain details about the behavior from parents, teachers, or other caregivers. Reasons for problem behaviors may include sensory reinforcement, task avoidance, boredom, and an attempt to communicate. A psychologist or behaviorist may design a program to decrease specific behaviors, which may include increasing the child's communication when possible. Medications may be helpful, including tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), alpha-2 agonists, benzodiazepines, seizure medications, and atypical antipsychotics. Consultation with a child psychiatrist is recommended if the medical home clinician is uncomfortable using these medications. Questions in the "Ask the Specialist" regarding Behavior in Children with CdLS (CdLS Foundation) may be helpful for providers and families. Behavioral modification can be extremely helpful, and consultation with a behavioral psychologist is recommended if behaviors escalate.
Specialty Collaborations & Other Services
Behavioral help is often necessary for children with CdLS. Depending on local expertise, developmental pediatrics and/or psychiatry may be helpful.
Psychiatry/Medication Management (see NW providers [0])
Developmental - Behavioral Pediatrics (see NW providers [1])
Sleep
- Consider physical causes of disrupted sleep, such as constipation, obstructive sleep apnea, and pain from gastroesophageal reflux and/or ear and sinus infections.
- Consider poor sleep hygiene (total sleep time, time to go to bed and to rise, quietness of the room, etc.) and suggest behavioral interventions. It might be helpful to ask the family to record sleep difficulties for a week or 2 to see the overall sleep pattern.
Specialty Collaborations & Other Services
Sleep Disorders (see NW providers [0])
Neurology
Maturation/Sexual/Reproductive
Specialty Collaborations & Other Services
Gynecology: Pediatric/Adolescent; Special Needs (see NW providers [0])
Eyes/Vision
Specialty Collaborations & Other Services
Pediatric Ophthalmology (see NW providers [1])
Ears/Hearing
Specialty Collaborations & Other Services
Pediatric Otolaryngology (ENT) (see NW providers [1])
Audiology (see NW providers [3])
Dental
Specialty Collaborations & Other Services
Pediatric Dentistry (see NW providers [2])
Ask the Specialist
My patient has no history or physical signs of gastroesophageal reflux and growth is appropriate for the CdLS growth charts. Would I still need to obtain a GI work-up for reflux?
Because reflux is so common in CdLS (80-90%), it is recommended that all individuals diagnosed with CdLS undergo at least an upper GI (gastrointestinal) series, if not an EGD (esophagogastroduodenoscopy), to rule out hidden reflux. Reflux can occur after it has been initially ruled out, and symptoms suggestive of reflux also should prompt a work-up.
In general, what is the minimum medical work-up to do after diagnosis of CdLS?
An echocardiogram, renal ultrasound, upper GI series to assess for reflux and also to rule out malrotation, pediatric ophthalmology evaluation, and audiology testing are generally recommended; most individuals should also have CdLS specific molecular testing.
Are prostheses recommended for individuals with CdLS without digits or with abnormal forearms?
Most individuals with absent digits and/or ulnar bones learn to manipulate everything with the few digits they have, and most do not seem to benefit greatly from hand and/or forearm prostheses. That said, a few individuals have used these with great success. Families considering their options may connect with others through the Cornelia de Lange Syndrome Foundation.
What are the typical causes of death in CdLS?
In the newborn period, these would include diaphragmatic hernia and/or severe congenital heart disease. Most children with CdLS live to adulthood, although a small percentage die earlier due to (in order of frequency): respiratory causes including aspiration/reflux and pneumonia, gastrointestinal disease including obstruction/volvulus, and congenital anomalies such as diaphragmatic hernia and heart defects. [Schrier: 2011]
Resources for Clinicians
On the Web
Information for Professionals (CdLS Foundation)
Materials addressing common issues and challenges related to CdLS - available at no cost to professionals. Additionally, professionals
can call and speak with a Family Service Coordinator or use an Ask the Expert form; Cornelia de Lange Syndrome Foundation.
Cornelia de Lange Syndrome (GeneReviews)
Detailed information addressing clinical characteristics, diagnosis/testing, management, genetic counseling, and molecular
pathogenesis; from the University of Washington and the National Library of Medicine.
Helpful Articles
PubMed search for Cornelia de Lange syndrome in children, last 4 years.
Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, Bengani H, Chan CY, et al.
Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of
mosaicism.
J Med Genet.
2014;51(10):659-68.
PubMed abstract / Full Text
Basel-Vanagaite L, Wolf L, Orin M, Larizza L, Gervasini C, Krantz ID, Deardoff MA.
Recognition of the Cornelia de Lange Syndrome Phenotype with Facial Dysmorphology Novel Analysis.
Clin Genet.
2015.
PubMed abstract
Cochran L, Moss J, Nelson L, Oliver C.
Contrasting age related changes in autism spectrum disorder phenomenology in Cornelia de Lange, Fragile X, and Cri du Chat
syndromes: Results from a 2.5 year follow-up.
Am J Med Genet C Semin Med Genet.
2015;169(2):188-97.
PubMed abstract
Moss J, Penhallow J, Ansari M, Barton S, Bourn D, FitzPatrick DR, Goodship J, Hammond P, Roberts C, Welham A, Oliver C.
Genotype-phenotype correlations in Cornelia de Lange syndrome: Behavioral characteristics and changes with age.
Am J Med Genet A.
2017;173(6):1566-1574.
PubMed abstract
Clinical Tools
Clinical Checklists & Visit Tools
Management and Treatment Guidelines (CdLS Foundation) ( 44 KB)
Highlights routine care for people with CdLS at different ages, including infancy, early and late childhood, adolescence,
and adulthood.
Developmental Skills Chart (CdLS Foundation) ( 98 KB)
A chart of the reported ages of completion for gross motor, fine motor, personal/social, and language milestones in children
with CdLS ages 1 month to 12 years; Cornelia de Lange Syndrome Foundation.
Growth/BMI Charts
Growth Charts for Boys (CdLS Foundation) ( 874 KB)
Since children with CdLS are often compared to a typical child’s growth rate, many
are incorrectly diagnosed with “failure to thrive,” a condition that focuses primarily on weight gain, so CdLS Foundation
Medical Director Antonie D. Kline, MD, and her colleagues, developed gender-specific growth charts based on research and
data collected over many years.
Growth Charts for Girls (CdLS Foundation) ( 1.4 MB)
Since children with CdLS are often compared to a typical child’s growth rate, many are incorrectly diagnosed with “failure
to thrive,” a condition that focuses primarily on weight gain, so CdLS Foundation Medical Director Antonie D. Kline, MD,
and her colleagues, developed gender-specific growth charts based on research and data collected over many years.
Resources for Patients & Families
Information on the Web
Cornelia de Lange Syndrome (MedlinePlus)
Information for families that includes description, frequency, causes, inheritance, other names, and additional resources;
from the National Library of Medicine.
Genetic Testing (CdLS Foundation)
A list of facilities that offer genetic testing for CdLS.
Spotlight on Cornelia de Lange Syndrome (PBS)
A compelling, 5-minute video about CdLS featured on Public Television stations nationwide.
National & Local Support
Support (CdLS Foundation)
Provides extensive information and support, including outreach programs and social workers to help families connect with local
resources.
Services for Patients & Families Nationwide (NW)
| Service Categories | # of providers* in: | NW | Partner states (4) (show) | | NM | NV | RI | UT | |
|---|---|---|---|---|---|---|---|---|---|
| Adolescent Health Transition Programs | 1 | 5 | 1 | 1 | 5 | ||||
| Audiology | 3 | 22 | 8 | 24 | 22 | ||||
| Cornelia de Lange Syndrome Clinics | |||||||||
| Developmental - Behavioral Pediatrics | 1 | 2 | 3 | 12 | 9 | ||||
| Dieticians and Nutritionists | 1 | 1 | 4 | 3 | 6 | ||||
| Early Intervention for Children with Disabilities/Delays | 3 | 34 | 30 | 13 | 51 | ||||
| General Counseling Services | 1 | 10 | 213 | 30 | 298 | ||||
| Gynecology: Pediatric/Adolescent; Special Needs | 3 | 9 | |||||||
| Medical Genetics | 1 | 2 | 5 | 4 | 7 | ||||
| Pediatric Dentistry | 2 | 6 | 24 | 54 | 50 | ||||
| Pediatric Endocrinology | 1 | 4 | 6 | 12 | 7 | ||||
| Pediatric Gastroenterology | 2 | 5 | 18 | 2 | |||||
| Pediatric Neurology | 5 | 5 | 18 | 8 | |||||
| Pediatric Ophthalmology | 1 | 6 | 6 | 8 | 4 | ||||
| Pediatric Orthopedics | 4 | 7 | 8 | 16 | 10 | ||||
| Pediatric Otolaryngology (ENT) | 1 | 11 | 5 | 7 | 10 | ||||
| Psychiatry/Medication Management | 3 | 37 | 80 | 53 | |||||
| Sleep Disorders | 2 | 1 | |||||||
| Special Education/Schools | 3 | 82 | 9 | 35 | 43 | ||||
| Speech - Language Pathologists | 4 | 23 | 11 | 34 | 65 | ||||
For services not listed above, browse our Services categories or search our database.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited to web-based services, provider locator services, and organizations that serve children from across the nation.
Bibliography
Allanson JE, Hennekam RC, Ireland M.
De Lange syndrome: subjective and objective comparison of the classical and mild phenotypes.
J Med Genet.
1997;34(8):645-50.
PubMed abstract / Full Text
Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, Bengani H, Chan CY, et al.
Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of
mosaicism.
J Med Genet.
2014;51(10):659-68.
PubMed abstract / Full Text
Avgitidou G, Cursiefen C, Heindl LM.
[Ophthalmological manifestations of Cornelia de Lange syndrome: Case report and review of the literature].
Ophthalmologe.
2015;112(5):455-8.
PubMed abstract
Barisic I, Tokic V, Loane M, Bianchi F, Calzolari E, Garne E, Wellesley D, Dolk H.
Descriptive epidemiology of Cornelia de Lange syndrome in Europe.
Am J Med Genet A.
2008;146A(1):51-9.
PubMed abstract
Basel-Vanagaite L, Wolf L, Orin M, Larizza L, Gervasini C, Krantz ID, Deardoff MA.
Recognition of the Cornelia de Lange Syndrome Phenotype with Facial Dysmorphology Novel Analysis.
Clin Genet.
2015.
PubMed abstract
Basile E, Villa L, Selicorni A, Molteni M.
The behavioural phenotype of Cornelia de Lange Syndrome: a study of 56 individuals.
J Intellect Disabil Res.
2007;51(Pt 9):671-81.
PubMed abstract
Cochran L, Moss J, Nelson L, Oliver C.
Contrasting age related changes in autism spectrum disorder phenomenology in Cornelia de Lange, Fragile X, and Cri du Chat
syndromes: Results from a 2.5 year follow-up.
Am J Med Genet C Semin Med Genet.
2015;169(2):188-97.
PubMed abstract
Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E,
Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T,
Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems
PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K.
HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle.
Nature.
2012;489(7415):313-7.
PubMed abstract / Full Text
Deardorff M A, Noon S E, Krantz I D.
Cornelia de Lange Syndrome.
GeneReviews [Internet]; (2016)
https://www.ncbi.nlm.nih.gov/books/NBK1104/. Accessed on Feb 15, 2019.
Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, Mönnich M, Yan Y, Xu W, Gil-Rodríguez MC, Clark
D, Hakonarson H, Halbach S, Michelis LD, Rampuria A, Rossier E, Spranger S, Van Maldergem L, Lynch SA, Gillessen-Kaesbach
G, Lüdecke HJ, Ramsay RG, McKay MJ, Krantz ID, Xu H, Horsfield JA, Kaiser FJ.
RAD21 mutations cause a human cohesinopathy.
Am J Hum Genet.
2012;90(6):1014-27.
PubMed abstract / Full Text
Dorsett D, Krantz ID.
On the molecular etiology of Cornelia de Lange syndrome.
Ann N Y Acad Sci.
2009;1151:22-37.
PubMed abstract
Kline AD, Krantz ID, Sommer A, Kliewer M, Jackson LG, FitzPatrick DR, Levin AV, Selicorni A.
Cornelia de Lange syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance.
Am J Med Genet A.
2007;143A(12):1287-96.
PubMed abstract
Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, Ishman SL, Kerr LM, Levin AV, Mulder PA, Ramos FJ,
Wierzba J, Ajmone PF, Axtell D, Blagowidow N, Cereda A, Costantino A, Cormier-Daire V, FitzPatrick D, Grados M, Groves L,
Guthrie W, Huisman S, Kaiser FJ, Koekkoek G, Levis M, Mariani M, McCleery JP, Menke LA, Metrena A, O'Connor J, Oliver C, Pie
J, Piening S, Potter CJ, Quaglio AL, Redeker E, Richman D, Rigamonti C, Shi A, Tümer Z, Van Balkom IDC, Hennekam RC.
Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement.
Nat Rev Genet.
2018.
PubMed abstract
Liu J, Krantz ID.
Cornelia de Lange syndrome, cohesin, and beyond.
Clin Genet.
2009;76(4):303-14.
PubMed abstract
McNairn AJ, Gerton JL.
Cohesinopathies: One ring, many obligations.
Mutat Res.
2008;647(1-2):103-11.
PubMed abstract
Moss J, Penhallow J, Ansari M, Barton S, Bourn D, FitzPatrick DR, Goodship J, Hammond P, Roberts C, Welham A, Oliver C.
Genotype-phenotype correlations in Cornelia de Lange syndrome: Behavioral characteristics and changes with age.
Am J Med Genet A.
2017;173(6):1566-1574.
PubMed abstract
Mulder PA, Huisman SA, Hennekam RC, Oliver C, van Balkom ID, Piening S.
Behaviour in Cornelia de Lange syndrome: a systematic review.
Dev Med Child Neurol.
2017;59(4):361-366.
PubMed abstract / Full Text
Olley G, Ansari M, Bengani H, Grimes GR, Rhodes J, von Kriegsheim A, Blatnik A, Stewart FJ, Wakeling E, Carroll N, Ross A,
Park SM, Bickmore WA, Pradeepa MM, FitzPatrick DR.
BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome.
Nat Genet.
2018;50(3):329-332.
PubMed abstract
Opitz JM.
The Brachmann-de Lange syndrome.
Am J Med Genet.
1985;22(1):89-102.
PubMed abstract
Parenti I, Gervasini C, Pozojevic J, Graul-Neumann L, Azzollini J, Braunholz D, Watrin E, Wendt KS, Cereda A, Cittaro D, Gillessen-Kaesbach
G, Lazarevic D, Mariani M, Russo S, Werner R, Krawitz P, Larizza L, Selicorni A, Kaiser FJ.
Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping
phenotype.
Clin Genet.
2016;89(1):74-81.
PubMed abstract
Pearce PM, Pitt DB.
Six cases of de Lange's syndrome; parental consanguinity in two.
Med J Aust.
1967;1(10):502-6.
PubMed abstract
Rajan R, Benke JR, Kline AD, Levy HP, Kimball A, Mettel TL, Boss EF, Ishman SL.
Insomnia in Cornelia de Lange syndrome.
Int J Pediatr Otorhinolaryngol.
2012;76(7):972-5.
PubMed abstract
Schrier SA, Sherer I, Deardorff MA, Clark D, Audette L, Gillis L, Kline AD, Ernst L, Loomes K, Krantz ID, Jackson LG.
Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of
the literature.
Am J Med Genet A.
2011;155A(12):3007-24.
PubMed abstract / Full Text
Symonds JD, Joss S, Metcalfe KA, Somarathi S, Cruden J, Devlin AM, Donaldson A, DiDonato N, Fitzpatrick D, Kaiser FJ, Lampe
AK, Lees MM, McLellan A, Montgomery T, Mundada V, Nairn L, Sarkar A, Schallner J, Pozojevic J, Parenti I, Tan J, Turnpenny
P, Whitehouse WP, Zuberi SM.
Heterozygous truncation mutations of the SMC1A gene cause a severe early onset epilepsy with cluster seizures in females:
Detailed phenotyping of 10 new cases.
Epilepsia.
2017;58(4):565-575.
PubMed abstract